THE INDIANA UNIVERSITY LIBRARY in Bloomington, IN is a stocky, ten-storey building where windows are a rarity.

In the dusk and dust of the tenth floor, one day in summer 2004, I was lucky enough to find the full series of the Proceedings of the Carlsberg Laboratories. I was looking for a rather unusual reference:
Winge O., 1923 On sex chromosomes, sex determination and preponderance of females in some dioecious plants. C. R. Trav. Lab. Carlsberg 15: 1–26.
When I eventually found it, it felt like Indiana Jones grasping the long-sought talisman.

Why on earth would I need such an esoteric paper?
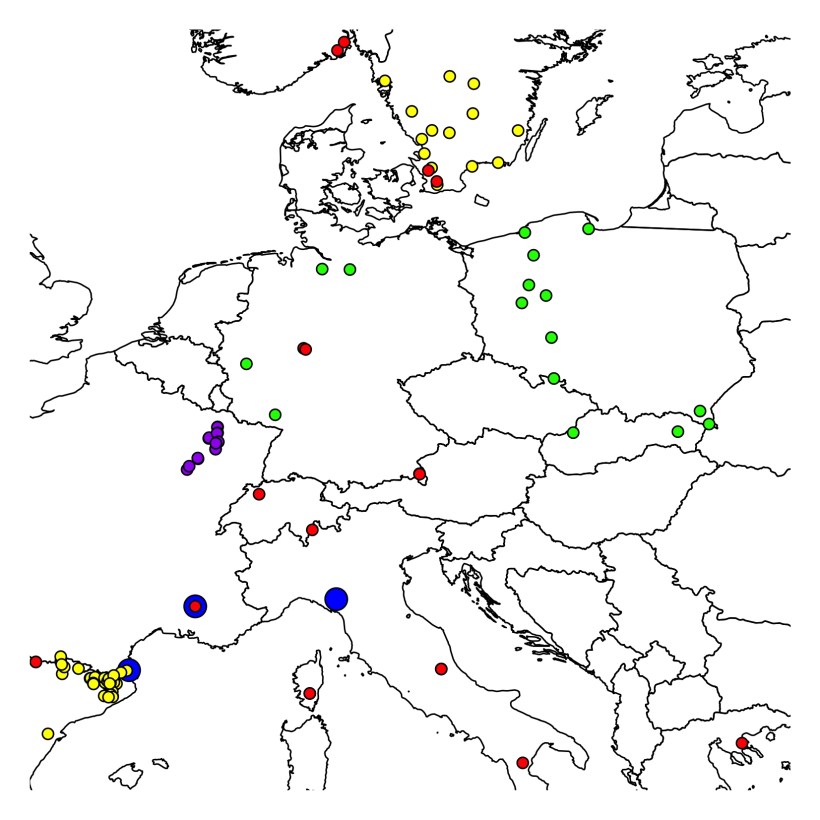
Few weeks earlier, as a post-doc in Lynda Delph’s lab (a very happy time indeed), I was doing linkage mapping with AFLP markers (sounds like the Stone Age, doesn’t it?) in Silene latifolia. The data, produced by Michele Arntz, were nice and tidy, and the map was coming out beautifully. Statistical support was excellent – actually, I’ve never seen such enormous LOD-scores, after or before then – so I was 100% confident in the map.

But something was wrong. ALL the literature on the species, which happens to be dioecious (i.e. it has separate sexes; the species, not the literature), claimed that, as in humans, the two sex chromosomes recombine only at one end (details vary, but all articles agreed on the one-end-only crossing overs).
And I had two nice recombinant blocks, one on each side of the non-recombining region. Oops. Ouch.
We went through all the analyses again, checked the data, the samples, re-ran the mapping with subsets of the mapping population. No way. The data resisted our efforts to force them to conform to the literary, consensus evidence.

It was time to ask for expert advice. So we called the best expert we could reach, a scientist with a very long experience in all matters plant sex chromosomes (let us treat this scientist as an anonymous referee, and let us not reveal her identity). She listened to us carefully, then after a pause she said: “but that’s… HERETIC!”.
I still consider this sentence as the best professional compliment I have ever received. But this did not push us forward an inch. We still had a blatant contradiction between our data and what science expected them to say, and nobody to explain why. Then Lynda, with her typical matter-of-factly, rigorous approach to science, decided we should go through the literature. ALL the literature on the subject.
Everybody quoted the 1923 Winge paper and several papers by Westergaard, published in the 40’s and in the 50’s (Westergaard (1946), Hereditas 32: 419-443; Westergaard (1948), Hereditas 34: 257-279; Westergaard (1958), Advances in Genetics 9: 217-281). All more recent literature quoted those papers as saying that “Silene latifolia sex chromosomes recombine at one end only”. Clearly, if there was a confrontation to be had, it was between our data and those papers.
Hence my travel in space and time to the last floor of IU library (Westergaard’s papers were easier to obtain).
The reading of those papers revealed the complex and surprising truth. Winge did not say a word about chiasmata patterns. Westergaard had carefully characterised the chiasmata on the sex chromosomes in the 1946 paper, but some data were hard to interpret, and so he chose to describe only “unusually favourable [mitotic] plates” (what we would call today “cherrypicking”, even though I acknowledge his great honesty in declaring it). Plus, he dismissed some chiasmata observed in the supposed “differential” arms as being “rare”. And went on describing differential and homologous arms of X and Y chromosomes.
Then, fatally, he depicted in Figure 5 a summary showing X and Y chromosomes with only one homologous arm, as in the figure above, the upper pane of which is based on his one. Westergaard, by all means, was a rigorous scientist, but he probably oversimplified his results in that drawing, and there we go: starting with this figure (and if you do not read the paper), you are dead sure that only one side recombines. Yet there was no contradiction between the data in the paper and our data. Unfortunately, following authors likely “quoted” only that figure from the 1946 paper. The result: against evidence, only one arm recombined now. Period.
This is serious. Such things should never happen in science. Certainly, most scientists read most of the papers they quote most of the time (for sure, compared to many politicians and opinion makers, we are diamond-grade examples of intellectual rigour, to be honest); but this story tells me that one must be suspicious when everybody quotes a very old, often hard to access, article (I bet very few people have read many of the Sewall Wright’s papers they quote, for instance; of the few who have read them, most have not understood a line of them; I belong to the latter category).
So, this is my take-home message from this story: read carefully all the papers you quote. If you cannot read them, then quote some other, more recent paper (e.g. a review), not the original message. In this case, the Chinese whispers chain must be explicit, if it has to exist at all (which I’d rather prefer not, anyway). Let us not convert science into a matter of ipse dixit. Let us stick to facts.
P.S. we actually published our linkage map, with sex chromosomes nicely recombining at both ends, here and here.








 thstanding its importance, genomic resources and a solid knowledge of the genomic bases of adaptation are lacking for the species.
thstanding its importance, genomic resources and a solid knowledge of the genomic bases of adaptation are lacking for the species.